Welcome to the EPISOL Colab Playground!
Solvation Free Energy: From SMILES strings to FEP-levels of Accuracy in Minutes¶

The 3D reference interaction site model (3DRISM) provides an efficient grid-based solvation model to compute the structural and thermodynamic properties of biomolecules in aqueous solutions, in this notebook we will walk through a high-throughput calculation on the solvation free energy of a test set of small molecules. We will compare our 3DRISM results to those of experimentally and computationally (FEP) determined free energies.
goals:
Generate the coordinate and topology files for our test set using RDKit and openFF
Perform high-throughput 3DRISM calculations to determine solvation free energy of 100 molecules in the test set
Be able to perform similar calculations on your own molecules
use the colab-notebook to download and run calculations on your own molecules
[ ]:
#@title ##Download and Install Episol
#@markdown ($\approx 2$min) Stable as of 07/01/25 eprism v1.2.6
%%capture
import subprocess
import pandas as pd
import matplotlib.pyplot as plt
#%cd ../home/
%cd $HOME/
%mkdir episol
%cd episol
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/fftw/fftw-3.3.8.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded fftw files"
!echo "+++++++++++++++++++"
!tar -xzf fftw-3.3.8.tar.gz
%cd fftw-3.3.8/
#!./configure --prefix=/home/fftw-3.3.8
!./configure --prefix=$HOME/episol/fftw-3.3.8
!make
!make install
%cd ../
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/kernel/release.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded Episol files"
!echo "+++++++++++++++++++"
!tar -xzf release.tar.gz
%cd release/
#!./configure --with-fftw=/home/fftw-3.3.8
!./configure --with-fftw=$HOME/episol/fftw-3.3.8
!make
!make install
#%cd /content
########################### WRAPEPR
import subprocess
import os
import threading
import pandas as pd
import matplotlib.pyplot as plt
!pip install episol
[ ]:
%%capture
#@title Install some python packages for topology generation
#@markdown ($\approx$4min)
#@markdown This will prompt a restart in our colab session, this is necessary, just keep moving
#@markdown (if you are using the notebook offline this wont be necessary, as presumably you'll have your own forcefield to generate topologies)
########################################
# FOR COLAB USERS ONLY #
#---------------------------------------#
# if you are running locally you dont need
# condacolab. Just use your local conda dist
########################################
!pip install -q condacolab
import condacolab
condacolab.install()
########################################
#!conda update conda
#!conda install --yes -c conda-forge python=3.11 numpy=1.26.4 openmm pdbfixer parmed mdanalysis py3dmol rdkit openff-toolkit
#!conda install -y -c conda-forge numpy=1.26.4 openmm=8.3.1 python={PYTHON_VERSION} pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 libgcc
!conda install -y -c conda-forge python=3.12 numpy=1.26.4 openmm=8.3.1 pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 torchvision
#openmm pdbfixer parmed mdanalysis py3dmol rdkitconda install libgcc
[ ]:
# FIX IMPORTER ERROR
!python -m ensurepip --upgrade
!pip3 install episol
!python3.12 -m pip install --upgrade setuptools
Looking in links: /tmp/tmp14g4fd4o
Requirement already satisfied: pip in /usr/local/lib/python3.12/site-packages (25.0.1)
WARNING: Running pip as the 'root' user can result in broken permissions and conflicting behaviour with the system package manager, possibly rendering your system unusable. It is recommended to use a virtual environment instead: https://pip.pypa.io/warnings/venv. Use the --root-user-action option if you know what you are doing and want to suppress this warning.
Requirement already satisfied: episol in /usr/local/lib/python3.12/site-packages (0.0.9)
Requirement already satisfied: setuptools in /usr/local/lib/python3.12/site-packages (80.9.0)
[ ]:
#@title import our download packages
%%capture
import py3Dmol
def MolTo3DView(mol, size=(300, 300), style="stick", surface=False, opacity=0.5):
"""
https://birdlet.github.io/2019/10/02/py3dmol_example/
Draw molecule in 3D
Args:
----
mol: rdMol, molecule to show
size: tuple(int, int), canvas size
style: str, type of drawing molecule
style can be 'line', 'stick', 'sphere', 'carton'
surface, bool, display SAS
opacity, float, opacity of surface, range 0.0-1.0
Return:
----
viewer: py3Dmol.view, a class for constructing embedded 3Dmol.js views in ipython notebooks.
"""
assert style in ('line', 'stick', 'sphere', 'carton')
mblock = Chem.MolToMolBlock(mol)
viewer = py3Dmol.view(width=size[0], height=size[1])
viewer.addModel(mblock, 'mol')
viewer.setStyle({style:{}})
if surface:
viewer.addSurface(py3Dmol.SAS, {'opacity': opacity})
viewer.zoomTo()
return viewer
def smi2conf(smiles):
'''Convert SMILES to rdkit.Mol with 3D coordinates'''
mol = Chem.MolFromSmiles(smiles)
if mol is not None:
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol, maxIters=200)
return mol
else:
return None
#free_energy("EXP")
!python -m ensurepip --upgrade # since we are using python 3.12 some pkg utils are now obsolete
# after conda-initiate restart colab resets pip
import matplotlib.pyplot as plt
import openmm as mm
from openmm import app
from openmm.unit import *
import py3Dmol as pymol
import MDAnalysis as md
import parmed as chem
from openff.toolkit.topology import Molecule, Topology
import numpy as np
from MDAnalysis.transformations import center_in_box
from episol import epipy
from rdkit import Chem
from rdkit.Chem import AllChem
from openff.toolkit.topology import Molecule
from openff.toolkit.utils import get_data_file_path
from openff.toolkit.typing.engines.smirnoff import ForceField
from openff.interchange import Interchange
# FIX IMPORTER ERROR
!python -m ensurepip --upgrade
!pip3 install episol
!python3.12 -m pip install --upgrade setuptools
%cd /content/
#Walk Through Calculation:
for this tutorial we will look at the solvation free energy of small molecules using FreeSolv Database
lets download our files
[ ]:
!wget https://github.com/MobleyLab/FreeSolv/raw/refs/heads/master/database.txt
--2025-09-08 00:02:23-- https://github.com/MobleyLab/FreeSolv/raw/refs/heads/master/database.txt
Resolving github.com (github.com)... 140.82.113.4
Connecting to github.com (github.com)|140.82.113.4|:443... connected.
HTTP request sent, awaiting response... 302 Found
Location: https://raw.githubusercontent.com/MobleyLab/FreeSolv/refs/heads/master/database.txt [following]
--2025-09-08 00:02:23-- https://raw.githubusercontent.com/MobleyLab/FreeSolv/refs/heads/master/database.txt
Resolving raw.githubusercontent.com (raw.githubusercontent.com)... 185.199.108.133, 185.199.109.133, 185.199.110.133, ...
Connecting to raw.githubusercontent.com (raw.githubusercontent.com)|185.199.108.133|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 144897 (142K) [text/plain]
Saving to: ‘database.txt’
database.txt 100%[===================>] 141.50K --.-KB/s in 0.03s
2025-09-08 00:02:23 (4.45 MB/s) - ‘database.txt’ saved [144897/144897]
now we will make a list containing allof the SMILES strings and their corresponding energies
[ ]:
line_count = int()
experimental_values = []
calculated_values = []
smiles_list = []
names_list = []
with open('database.txt','r') as r:
for line in r:
line_count +=1
if line_count > 3:
tmp = line.split(';')
try:
names_list.append('_'.join(tmp[2].split()))
smiles_list.append(tmp[1].strip())
experimental_values.append(float(tmp[3].strip()))
calculated_values.append(float(tmp[5].strip()))
except Exception as exc:
RuntimeWarning(exc)
smiles_list = np.array(smiles_list)
names_list = np.array(names_list)
experimental_values = np.array(experimental_values)
calculated_values = np.array(calculated_values)
lets look at one of our (randomly picked) molecules
[ ]:
rng = np.random.default_rng()
ind_to_extract = rng.integers(len(smiles_list),size=1)[0]
test = smiles_list[ind_to_extract]
mol = Chem.MolFromSmiles(test)
mol = Chem.rdmolops.AddHs(mol,addCoords=True)
mol
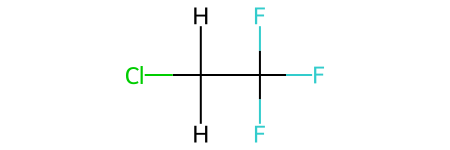
we can see our molecule has totally non-physical geometry
lets preform a simple geometry optimizationn step and view the output
[ ]:
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
mol
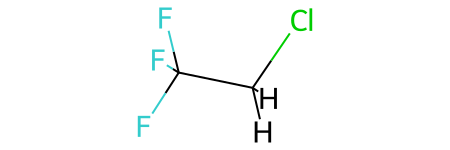
much better
now lets investigate solvation free energy of our molecule
First we need to make our topology file
thankfully using openFF and SMIRNOFF we can do this no problem!
[ ]:
#!python -m ensurepip --upgrade
# use openFF..toolkit to accept rdkit object
from_rdmol = Molecule.from_rdkit(mol,allow_undefined_stereo=True)
topology = from_rdmol.to_topology()
# we will use openFF to assign SMIRNOFF parameters
sage = ForceField("openff-2.0.0.offxml")
interchange = Interchange.from_smirnoff(force_field=sage, topology=topology)
# just pick the first conformer
interchange.positions = from_rdmol.conformers[0]
#openmm_system = interchange.to_gromacs('out')
openmm_system = interchange.to_openmm()
#os.remove("out.top")
interchange.to_top("example.top")
tmp_u = md.Universe(mol)
coords = tmp_u.atoms.positions
# Buffer will be greater than 1nm (our default cutoff)
# here we set to 7A on each side, so at least 1.4 nm apart from the other side of
# the molecule
buffer = 5 # convert to A
#
box_x = np.ceil(np.abs(np.max((coords[:,0]))-np.min((coords[:,0])))+buffer)
box_y = np.ceil(np.abs(np.max((coords[:,1]))-np.min((coords[:,1])))+buffer)
box_z = np.ceil(np.abs(np.max((coords[:,2]))-np.min((coords[:,2])))+buffer)
tmp_u.dimensions = [box_x,box_y,box_z,90,90,90]
trans = center_in_box(tmp_u.atoms,center='geometry')
tmp_u.trajectory.add_transformations(trans)
tmp_u.atoms.write(f'fixed_example.gro') # have to write out structure file
## run 3DRISM
test = epipy(f'fixed_example.gro','example.top',gen_idc=True)
test.ndiis = 15
test.delvv = 0.5
test.r_c = 0.9 # cutoff at 0.9 nm instead of 1 nm (default)
test.err_tol = 1e-12
test.rism(resolution=0.5)
test.kernel(nt=2)
# We will use epipy's automatic unit conversion
# to specifiy our free energy units
test.free_energy()
/usr/local/lib/python3.12/site-packages/openff/amber_ff_ports/amber_ff_ports.py:8: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import resource_filename
/usr/local/lib/python3.12/site-packages/MDAnalysis/coordinates/GRO.py:444: UserWarning: Supplied AtomGroup was missing the following attributes: resnames. These will be written with default values. Alternatively these can be supplied as keyword arguments.
warnings.warn(
converted example.top to example.solute
generated idc-enabled solute file to: idc_example.solute
Calculation finished in 233 steps
err_tol: 1e-12 actual: 9.38025e-13
0.8673300582465404
our output energy is in units of kJ/mol
we can specify the units if need be by feeding our desired string to the function
[ ]:
test.free_energy('kcal/mol')
0.20729685904554024
lets see the energy per molecuule
[ ]:
test.free_energy('kcal')
1.2483416851722435e+23
now lets run the first N molecules in the test set
[ ]:
number_of_molecules_to_use = 50
rng = np.random.default_rng()
ind_to_extract = rng.integers(len(smiles_list),size=number_of_molecules_to_use)
#smiles_and_names = dict(zip(names_list[ind_to_extract],smiles_list[ind_to_extract]))
smiles_and_names = dict(zip(names_list[:number_of_molecules_to_use],smiles_list[:number_of_molecules_to_use]))
[ ]:
len(smiles_and_names)
50
Run ! \(\approx\) 5min for the first 50
[ ]:
%%capture
#mols = [Chem.MolFromSmiles(x) for x in smiles_list[:5]]
def generate_topology_and_get_energy(smiles_list:list,keep_files=False):
"""
This will automatically generate topology files for
any small molecule SMILES-string.
!!! Warning !!!
We are using undefined stereochemistry
-----------
input: smiles_list -> list of SMILES strings
output: energy_list -> list of solvation free energies whose
index corresponds to the SMILES string input
keep_files: boolean -> do you want to save ALL file generated
or just keep the energy
"""
import os
out_energies = []
try:
for name in smiles_list:
stage = "convert to rdkit mol"
mol = Chem.MolFromSmiles(name)
mol = Chem.rdmolops.AddHs(mol,addCoords=True)
stage = "Add hydrogens and Geometry optimize "
# geometry optimization step
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
##
stage = "Pass to openFF "
# use openFF..toolkit to accept rdkit object
from_rdmol = Molecule.from_rdkit(mol,allow_undefined_stereo=True)
topology = from_rdmol.to_topology()
# we will use openFF to assign SMIRNOFF parameters
stage = "Use smirnoff "
sage = ForceField("openff-2.0.0.offxml")
interchange = Interchange.from_smirnoff(force_field=sage, topology=topology)
# just pick the first conformer
interchange.positions = from_rdmol.conformers[0]
#openmm_system = interchange.to_gromacs('out')
openmm_system = interchange.to_openmm()
#os.remove("out.top")
stage = "Generate topology"
interchange.to_top("out.top")
tmp_u = md.Universe(mol)
coords = tmp_u.atoms.positions
# Buffer will be greater than 1nm (our default cutoff)
# here we set to 7A on each side, so at least 1.4 nm apart from the other side of
# the molecule
buffer = 5 # convert to A
#
stage = "Center our molecule in a PBC"
box_x = np.ceil(np.abs(np.max((coords[:,0]))-np.min((coords[:,0])))+buffer)
box_y = np.ceil(np.abs(np.max((coords[:,1]))-np.min((coords[:,1])))+buffer)
box_z = np.ceil(np.abs(np.max((coords[:,2]))-np.min((coords[:,2])))+buffer)
tmp_u.dimensions = [box_x,box_y,box_z,90,90,90]
trans = center_in_box(tmp_u.atoms,center='geometry')
tmp_u.trajectory.add_transformations(trans)
tmp_u.atoms.write(f'fixed_mol.gro') # have to write out structure file
stage = "Start 3DRISM Calculation"
## run 3DRISM
test = epipy(f'fixed_mol.gro','out.top',gen_idc=True)
test.ndiis = 15
test.delvv = 0.5
test.r_c = 0.9 # cutoff at 0.9 nm instead of 1 nm (default)
test.err_tol = 1e-12
test.rism(resolution=0.5)
test.kernel(nt=2)
# We will use epipy's automatic unit conversion
# to specifiy our free energy units
out_energies.append(test.free_energy('kcal/mol'))
# get rid of our calculation files
# we dont really care about saving them
# this way we dont have files piling up
if not keep_files:
os.remove("out.top")
os.remove('fixed_mol_out.ts4s')
os.remove('fixed_mol_out.log')
except Exception as exc:
print(f"molecule: {name} failed at stage {stage}")
print(RuntimeError(exc))
return out_energies
# Now we run
out_energies = generate_topology_and_get_energy(smiles_list=[smiles for smiles in smiles_and_names.values()])
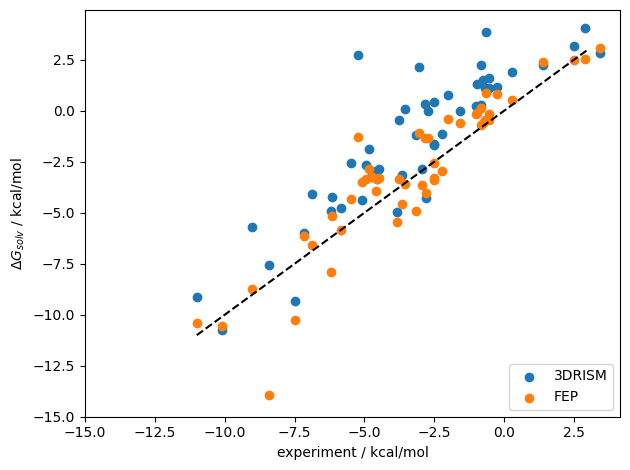
now, lets see our results
[ ]:
fig,ax = plt.subplots()
#out_energies = np.copy(np.array(out_en))
dslice = len(out_energies)
ax.scatter(experimental_values[:dslice],out_energies,label='3DRISM')
ax.scatter(experimental_values[:dslice],calculated_values[:dslice],label='FEP')
#ax1.scatter(calculated_values[:dslice],out_energies)
#ax1.set_xlabel("experimental values")
ax.set_ylabel("$\\Delta G_{solv}$ / kcal/mol")
ax.set_xlabel("experiment / kcal/mol")
vv = np.arange(min(experimental_values[:dslice]),max(experimental_values[:dslice]))
ax.plot(vv,vv,'k--')
ax.set_ylim(-15,None)
ax.set_xlim(-15,None)
ax.legend(loc="lower right")
fig.tight_layout()
feel free to use this notebook how you desire
run the whole dataset or upload your own!
we’ve included a section below where you can make your own molecule and run calculations
get creative!
[ ]:
smiles_string = "ClC=C=CCO" #@param {type: 'string'}
def smi2viewer(smi='CC=O'):
try:
conf = smi2conf(smi)
return MolTo3DView(conf).show()
except:
return None
smi2viewer(smi=smiles_string)
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
Run 3DRISM
[ ]:
print("your free energy of solvation is ",generate_topology_and_get_energy([smiles_string]),'kcal/mol')
/usr/local/lib/python3.12/site-packages/MDAnalysis/coordinates/GRO.py:444: UserWarning: Supplied AtomGroup was missing the following attributes: resnames. These will be written with default values. Alternatively these can be supplied as keyword arguments.
warnings.warn(
converted out.top to out.solute
generated idc-enabled solute file to: idc_out.solute
Calculation finished in 267 steps
err_tol: 1e-12 actual: 9.92814e-13
your free energy of solvation is [-5.00284805149884] kcal/mol