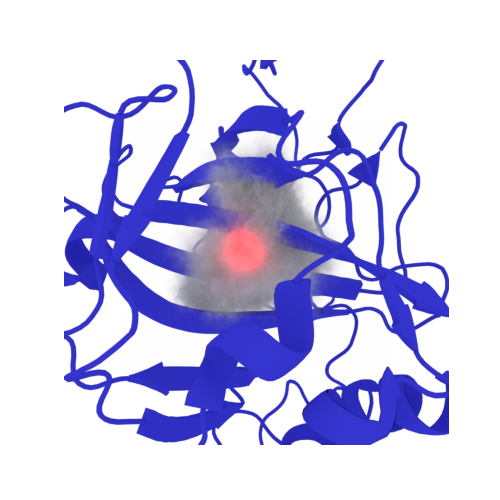
Have you ever needed a reliable way to place the initial positions of Mg²⁺ ions in RNA or DNA molecular dynamics simulations? These divalent ions play a critical role in nucleic acid structure and dynamics.
This tutorial demonstrating how to achieve this using our EPIPY package. The method is based on the 3DRISM-IDC theory, which was specifically developed to model solvation around highly negatively charged solutes such as nucleic acids.
If you use the materials in this tutorial, please cite: “A Python Tutorial for 3DRISM Solvation Calculations of Chemical and Biological Molecules”, Swanson, P., Cao, S., Huang, X, https://chemrxiv.org/engage/chemrxiv/article-details/68a6903c728bf9025e6c91ed
Placing Magnesium in Highly Correlated Regions of RNA kink-Turn¶
[1]:
#@title ##Download and Install Episol
#@markdown ($\approx 2$min) Stable as of 07/01/25 eprism v1.2.6
%%capture
import subprocess
import pandas as pd
import matplotlib.pyplot as plt
#%cd ../home/
%cd $HOME/
%mkdir episol
%cd episol
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/fftw/fftw-3.3.8.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded fftw files"
!echo "+++++++++++++++++++"
!tar -xzf fftw-3.3.8.tar.gz
%cd fftw-3.3.8/
#!./configure --prefix=/home/fftw-3.3.8
!./configure --prefix=$HOME/episol/fftw-3.3.8
!make
!make install
%cd ../
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/kernel/release.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded Episol files"
!echo "+++++++++++++++++++"
!tar -xzf release.tar.gz
%cd release/
#!./configure --with-fftw=/home/fftw-3.3.8
!./configure --with-fftw=$HOME/episol/fftw-3.3.8
!make
!make install
#%cd /content
########################### WRAPEPR
!pip install episol==0.1.2
#!python3.12 -m pip install --upgrade setuptools
import subprocess
import os
import threading
import pandas as pd
import matplotlib.pyplot as plt
from episol import epipy
[2]:
%%capture
#@title Install some python packages for topology generation
#@markdown ($\approx$4-8min)
#@markdown This will prompt a restart in our colab session, this is necessary, just keep moving
#@markdown (if you are using the notebook offline this wont be necessary, as presumably you'll have your own forcefield to generate topologies)
########################################
# FOR COLAB USERS ONLY #
#---------------------------------------#
# if you are running locally you dont need
# condacolab. Just use your local conda dist
########################################
!pip install -q condacolab
import condacolab
condacolab.install()
########################################
#!conda update conda
#!conda install --yes -c conda-forge python=3.11 numpy=1.26.4 openmm pdbfixer parmed mdanalysis py3dmol rdkit openff-toolkit
#!conda install -y -c conda-forge numpy=1.26.4 openmm=8.3.1 python={PYTHON_VERSION} pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 libgcc
!conda install -y -c conda-forge python=3.12 numpy=1.26.4 openmm=8.3.1 pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 torchvision pymol-open-source
!python3.12 -m pip install --upgrade setuptools
!python -m ensurepip --upgrade
#openmm pdbfixer parmed mdanalysis py3dmol rdkitconda install libgcc
[1]:
#@title import our downloaded packages
%%capture
!python -m ensurepip --upgrade # since we are using python 3.12 some pkg utils are now obsolete
# after conda-initiate restart colab resets pip
import matplotlib.pyplot as plt
import openmm as mm
from openmm import app
# fix later
from openmm.app import *
from openmm.unit import *
import py3Dmol# as pymol
import MDAnalysis as md
import parmed as chem
from openff.toolkit.topology import Molecule, Topology
import numpy as np
import MDAnalysis.transformations as mdt
import pdbfixer
from episol import epipy
%cd /content/
#Walk Through Calculation:
First, we will download our desired structure file from the PDB
[30]:
PDB_ID='7EFH' # @param {type:"string", placeholder:"enter a value"}
PDB_ID = PDB_ID.strip()
!wget https://files.rcsb.org/download/"{PDB_ID}.pdb"
--2025-10-28 21:21:58-- https://files.rcsb.org/download/7EFH.pdb
Resolving files.rcsb.org (files.rcsb.org)... 3.166.135.85, 3.166.135.84, 3.166.135.67, ...
Connecting to files.rcsb.org (files.rcsb.org)|3.166.135.85|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: unspecified [text/plain]
Saving to: ‘7EFH.pdb.2’
7EFH.pdb.2 [ <=> ] 53.47K --.-KB/s in 0.01s
2025-10-28 21:21:58 (4.11 MB/s) - ‘7EFH.pdb.2’ saved [54756]
we can see that our unit cell is very large, using such a large box as input for 3DRISM is possible, but the extra ‘blank space’ equates to an excessive ammout of RAM
to combat this we will modify the unit cell to be much smaller, saving cost while maintaining acuuracy
since we are using a interaction-cutoff of 1nm, we will add a buffer on our PBC cell
Again, before we run 3DRISM, we need to generate a topology file for our molecule
we will use PDBFixer to add missing residues, atoms and hydrogens
[31]:
buffer = 7 # angstroms
now we clean our PDB file and generate a topology, we wont include any ions in this example
this protocol will work for essentailly any PDB (within reason) so feel free to download a different PDB and redo the tutorial
[32]:
#@markdown Once again, lets clean our PDB
fixer = pdbfixer.PDBFixer(f'{PDB_ID}.pdb')
forcefield = app.ForceField('amber14-all.xml','amber14/tip3pfb.xml')#charmm36.xml')
buffer = 7
stage = "Finding missing residues..."
fixer.findMissingResidues()
stage = "Finding nonstandard residues..."
chains = list(fixer.topology.chains())
keys = fixer.missingResidues.keys()
keys = [i for i in fixer.missingResidues.keys()]
for key in keys:
chain = chains[key[0]]
if key[1] == 0 or key[1] == len(list(chain.residues())):
del fixer.missingResidues[key]
fixer.findNonstandardResidues()
#fixer.findNonstandardResidues()
stage = "Replacing nonstandard residues..."
fixer.replaceNonstandardResidues()
stage = "Removing heterogens..."
fixer.removeHeterogens(keepWater=False)
stage = "Finding missing atoms..."
fixer.findMissingAtoms()
stage = "Adding missing atoms..."
fixer.addMissingAtoms()
stage = "Adding missing hydrogens..."
fixer.addMissingHydrogens(pH=7)
#stage = "Writing PDB file..."
#app.PDBFile.writeFile(fixer.topology, fixer.positions, 'fixed_1ALU.pdb')
stage = "Create System..."
fixer = Modeller(fixer.topology,fixer.positions)
system = forcefield.createSystem(fixer.topology)
#fixer = Modeller(small.topology,small.positions)
struct = chem.openmm.load_topology(fixer.topology,system)
struct.positions = fixer.positions
struct.save(f'{PDB_ID}.top',overwrite=True)
####################### Pass to MDAnalysis and recenter box
temp = md.Universe(struct)#.select_atoms('all')
coords = temp.atoms.positions
"""
trans = [mdt.boxdimensions.set_dimensions([2*np.ceil(np.max(np.abs(coords[:,0]))+buffer),
2*np.ceil(np.max(np.abs(coords[:,1]))+buffer),
2*np.ceil(np.max(np.abs(coords[:,2]))+buffer),
90,90,90]),
mdt.center_in_box(temp.atoms,center='geometry')]
"""
box_x = np.ceil(np.abs(np.max((coords[:,0]))-np.min((coords[:,0])))+buffer)
box_y = np.ceil(np.abs(np.max((coords[:,1]))-np.min((coords[:,1])))+buffer)
box_z = np.ceil(np.abs(np.max((coords[:,2]))-np.min((coords[:,2])))+buffer)
temp.dimensions = [box_x,box_y,box_z,90,90,90]
trans = mdt.center_in_box(temp.atoms,center='geometry')
# we must multiply by 2 because all AF PDBS start at origin I_3*1
temp.trajectory.add_transformations(trans)
temp.atoms.write(f'fixed_{PDB_ID}.gro')
temp.atoms.write(f'fixed_{PDB_ID}.pdb') # py3Dmol can only use pdb unit cell
box,n_atoms,n_res = temp.dimensions[:3], len(temp.atoms), len(temp.residues)
#struct.save(f'fixed_{name}.gro',overwrite=True)
disp = py3Dmol.view()
disp.addModel(open(f'fixed_{PDB_ID}.pdb', 'r').read(),'pdb')
disp.setStyle({'model': -1}, {'cartoon': {}})
disp.addModel(open(f'fixed_{PDB_ID}.pdb', 'r').read(),'pdb')
disp.setStyle({'model': -1}, {'stick': {}})
disp.addUnitCell()
disp.zoomTo()
disp.show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
[33]:
pdb = epipy(f"fixed_{PDB_ID}.gro",f"{PDB_ID}.top",convert=True)
converted 7EFH.top to 7EFH.solute
Here we specifiy our solvent.
Our current options are ‘water’ or ‘MG’
however, “MG” includes water as well
[34]:
pdb.solvent("mg")
Here we can see our forcefield file for our solvent
[35]:
pdb.solvent_top
[35]:
'tip3p-MgCl-amber18-full.gaff'
as before, we set our saved file name to one relevant as opposed to default names
[36]:
pdb.report(f'{PDB_ID}')
When using solvent mixtures, we have found that the KGK or KH closure works best
for the scope of this tutorial will will use the KGK closure, however, readers aiming for more accurate results are recommended to use the KH closure
[37]:
pdb.rism(step=10000,resolution=1)
pdb.closure = "KGK"
pdb.err_tol = 1e-06 # we will set a higher tolerance than usual
now we run!
since we are using a bit of a larger solute, we will set our number of threads to 2, using the -nt flag
~ 5min
[38]:
#pdb.ndiis = 15
pdb.kernel(nt=2)
Calculation finished in 870 steps
err_tol: 1e-06 actual: 9.9199e-07
visualizing resutls
lets extract our atomic-density from the calculation
here we can just specify our atom (MG) we want to use
[39]:
g_r = pdb.select_grid('guv of atom mg')
we can now visualize our results
[40]:
z_slice = 24 # @param {type:"slider", min:1, max:91, step:1}
fig,ax = plt.subplots()
#z_slice = 10
p = ax.pcolormesh(g_r[:,:,z_slice])
ax.set_ylabel(f'y grid / {pdb.resolution}$\\mathring{{A}}$')
ax.set_xlabel(f'x grid / {pdb.resolution}$\\mathring{{A}}$')
#ax.set_ylim(20,100)
#ax.set_xlim(10,65)
fig.colorbar(p,ax=ax, label="$g(\\vec{r})/\\mathring{{A}}$")
fig.suptitle(f'$g(\\vec{{r}})_{{MG}}$ at slice {z_slice*pdb.resolution} $\\mathring{{A}}$')
[40]:
Text(0.5, 0.98, '$g(\\vec{r})_{MG}$ at slice 24 $\\mathring{A}$')
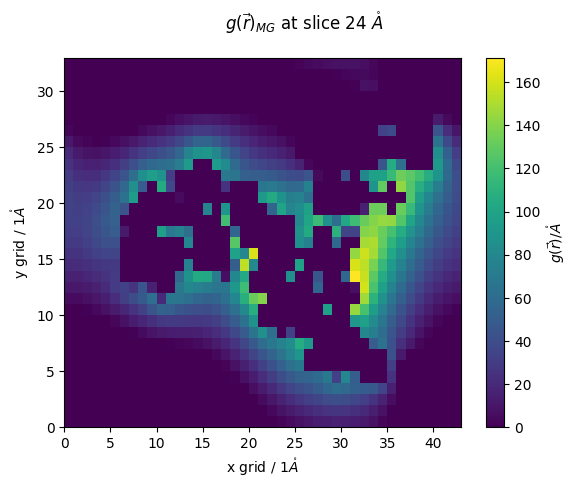
Now lets place ions on our grid
“number_mg_to_place = 5” - we chose to place 5 Mg2+ surronding RNA. You can choose this value per your understand of your system.
[41]:
number_mg_to_place = 5
placed_waters = pdb.placement(number_mg_to_place,atom_to_select='mg',write_pdb=True,outname=f'placed_mg_{PDB_ID}')#,filename='guv_diis_increase_pdb.txt',write_pdb=True,outname='placed_mg')
[42]:
number_mg_to_place = 5
placed_waters = pdb.placement(number_mg_to_place,atom_to_select='mg',write_pdb=True,
outname=f'placed_mg_{PDB_ID}')#,filename='guv_diis_increase_pdb.txt',write_pdb=True,outname='placed_mg')
[43]:
#@markdown Here we merge our two PDBs and write them to merged_pdb.pdb
%%capture
u1 = md.Universe(f'fixed_{PDB_ID}.gro')
u2 = md.Universe(f'placed_mg_{PDB_ID}.pdb')
#u2.dimensions = [il6.grid[0],il6.grid[1],il6.grid[2],90,90,90]
md.Merge(u1.atoms,u2.atoms).atoms.write(f'merged_pdb_{PDB_ID}.pdb')
[44]:
#@markdown View our placed Magnesium
#md.Universe("aligned_merged_pdb.pdb").select_atoms("resname HOH").atoms.write('aligned_waters.pdb')
#md.Universe(f"{PDB_ID}.pdb").select_atoms("resname HOH").atoms.write('orig_waters.pdb')
disp = py3Dmol.view()
disp.addModel(open(f'fixed_{PDB_ID}.pdb','r').read(),'pdb')
#disp.addModel(open('aligned_merged_pdb.pdb','r').read(),'pdb')
disp.setStyle({'model': -1}, {'cartoon': {'color':'grey'}})
#disp.addModel(open('aligned_waters.pdb', 'r').read(),'pdb')
disp.addModel(open(f'placed_mg_{PDB_ID}.pdb', 'r').read(),'pdb',)
#disp.setStyle({'model': -1}, {'sphere': {}})
disp.setStyle({'model': -1}, {'sphere': {'color':'purple'}})
#disp.addModel(open('orig_waters.pdb', 'r').read(),'pdb')
#disp.setStyle({'model': -1}, {'sphere': {'color':'red'}})
#disp.addUnitCell()
#disp.addLabel('Cyan: RISM , Red: True')#,"bottomCenter")#{'alignment':"bottomCenter"},{'fontColor':'cyan'})#:{'color':'cyan'}})#,{'color':'red'})
disp.setHoverable({},True,'''function(atom,viewer,event,container) {
if(!atom.label) {
atom.label = viewer.addLabel(atom.resn+":"+atom.atom,{position: atom, backgroundColor: 'mintcream', fontColor:'black'});
}}''',
'''function(atom,viewer) {
if(atom.label) {
viewer.removeLabel(atom.label);
delete atom.label;
}
}''')
disp.zoomTo()
disp.show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
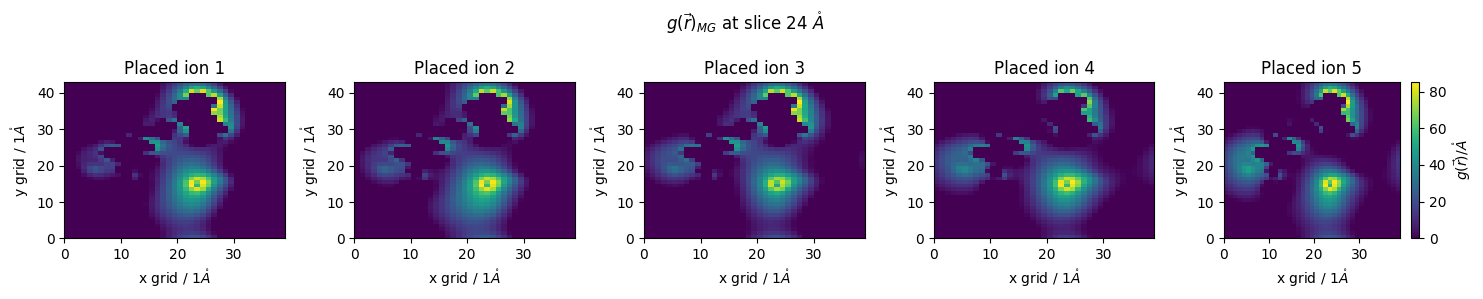
we can see that our placed ions are in close proximity
Our selection command relies on a greedy picking algorithm, picking the highest peak in the distribution, then moving on to the next
When placing highly charged solvents, one can imagine how placing a single ion, then moving to the next highest peak could be distruptive of the true behavior of the system, as regions of great distribution may be close to one another
Instead, we can place an ion, then redo our 3DRISM calcultaion
This is specifically important for highly-highly charged regions, for example kink-turn regions of RNA
[45]:
#@markdown Here we will redo a 3DRISM calculation each time we place an Ion
#@markdown $\approx$ 1min per iteration
mg_you_want = 5 #@param
out = []
pdb = epipy(f"fixed_{PDB_ID}.gro",f"{PDB_ID}.top",convert=True)
for mg in range(mg_you_want):
print(f"Starting round {mg}\n")
pdb.solvent("mg")
pdb.report(f"placed_mg_{mg}_{PDB_ID}")
#pdb.rism(resolution=0.5)
pdb.rism(step=10000,resolution=1)
pdb.closure = "KGK"
pdb.err_tol = 1e-06 # we will set a higher tolerance than usual
pdb.kernel(nt=2)
out.append(pdb.select_grid('guv of atom mg')[z_slice])
number_mg_to_place = 1 # per loop
placed_waters = pdb.placement(number_mg_to_place,atom_to_select='mg',write_pdb=True,outname=f'placed_mg_{mg}_{PDB_ID}')#,filename='guv_diis_increase_pdb.txt',write_pdb=True,outname='placed_mg')
print(f"finished round {mg}\n")
## now we are done with the calc
if mg == 0:
u1 = md.Universe(f"fixed_{PDB_ID}.gro")
else:
u1 = md.Universe(f'merged_pdb_{mg-1}_{PDB_ID}.pdb')
#
u2 = md.Universe(f'placed_mg_{mg}_{PDB_ID}.pdb') #this is the MG we just placed
# this wont be needed in epipy update
# but i wrote my pdb atomnames one column too far
u2.add_TopologyAttr('resnames',['Mg'])
u2.add_TopologyAttr('elements',['Mg'])
u2.add_TopologyAttr('names',['Mg'])
u2.add_TopologyAttr('resid',[mg])
# merge the two files
u3 = md.Merge(u1.atoms,u2.atoms)
# we have to reset the dimensions
u3.dimensions = [box_x,box_y,box_z,90,90,90]
# write out
u3.atoms.write(f'merged_pdb_{mg}_{PDB_ID}.pdb')
u3.atoms.write(f'merged_pdb_{mg}_{PDB_ID}.gro')
n_mg = len(u3.select_atoms("name Mg")) # get number of MG as failsafe
print(f"Merged the PDB and placed ion\nnow contains {n_mg} MG ions\n")
# now make a new topology
fixer = app.PDBFile(f'merged_pdb_{mg}_{PDB_ID}.pdb')
forcefield = app.ForceField('amber14-all.xml','amber14/tip3pfb.xml')#charmm36.xml')
system = forcefield.createSystem(fixer.topology)
# print the topology via RDKit
struct = chem.openmm.load_topology(fixer.topology,system)
struct.positions = fixer.positions
struct.save(f'{PDB_ID}_{mg}.top',overwrite=True)
# Read into epipy
pdb = epipy(f'merged_pdb_{mg}_{PDB_ID}.gro',f"{PDB_ID}_{mg}.top",convert=True)
converted 7EFH.top to 7EFH.solute
Starting round 0
Calculation finished in 870 steps
err_tol: 1e-06 actual: 9.9199e-07
finished round 0
Merged the PDB and placed ion
now contains 1 MG ions
converted 7EFH_0.top to 7EFH_0.solute
Starting round 1
Calculation finished in 904 steps
err_tol: 1e-06 actual: 9.8827e-07
finished round 1
Merged the PDB and placed ion
now contains 2 MG ions
converted 7EFH_1.top to 7EFH_1.solute
Starting round 2
Calculation finished in 851 steps
err_tol: 1e-06 actual: 9.96661e-07
finished round 2
Merged the PDB and placed ion
now contains 3 MG ions
converted 7EFH_2.top to 7EFH_2.solute
Starting round 3
Calculation finished in 948 steps
err_tol: 1e-06 actual: 9.93306e-07
finished round 3
Merged the PDB and placed ion
now contains 4 MG ions
converted 7EFH_3.top to 7EFH_3.solute
Starting round 4
Calculation finished in 774 steps
err_tol: 1e-06 actual: 9.90788e-07
finished round 4
Merged the PDB and placed ion
now contains 5 MG ions
converted 7EFH_4.top to 7EFH_4.solute
[46]:
#@markdown lets plot the density difference between iterations
fig,axs = plt.subplots(1,mg_you_want,figsize=(15,3))
#z_slice = 10
for idx,(out_i,ax) in enumerate(zip(out,axs.flatten())):
#p = ax.pcolormesh(g_r[:,:,z_slice])
p = ax.pcolormesh(out_i)
ax.set_ylabel(f'y grid / {pdb.resolution}$\\mathring{{A}}$')
ax.set_xlabel(f'x grid / {pdb.resolution}$\\mathring{{A}}$')
ax.set_title(f"Placed ion {idx+1}")
#ax.set_ylim(20,100)
#ax.set_xlim(10,65)
fig.colorbar(p,ax=ax, label="$g(\\vec{r})/\\mathring{{A}}$")
fig.suptitle(f'$g(\\vec{{r}})_{{MG}}$ at slice {z_slice*pdb.resolution} $\\mathring{{A}}$')
fig.tight_layout()
[47]:
#@markdown Now lets view the 'properly placed' Ions
u = md.Universe(f'merged_pdb_{mg}_{PDB_ID}.pdb')
u.select_atoms("name Mg").write("total_mg_placed.pdb")
disp = py3Dmol.view()
disp.addModel(open(f'merged_pdb_{mg}_{PDB_ID}.pdb','r').read(),'pdb')
#disp.addModel(open('aligned_merged_pdb.pdb','r').read(),'pdb')
disp.setStyle({'model': -1}, {'stick': {}})
##################
disp.addModel(open('total_mg_placed.pdb','r').read(),'pdb')
disp.setStyle({'model': -1}, {'sphere': {'color':'purple'}})
#disp.addModel(open('orig_waters.pdb', 'r').read(),'pdb')
#disp.setStyle({'model': -1}, {'sphere': {'color':'red'}})
#disp.addUnitCell()
#disp.addLabel('Cyan: RISM , Red: True')#,"bottomCenter")#{'alignment':"bottomCenter"},{'fontColor':'cyan'})#:{'color':'cyan'}})#,{'color':'red'})
disp.setHoverable({},True,'''function(atom,viewer,event,container) {
if(!atom.label) {
atom.label = viewer.addLabel(atom.resn+":"+atom.atom,{position: atom, backgroundColor: 'mintcream', fontColor:'black'});
}}''',
'''function(atom,viewer) {
if(atom.label) {
viewer.removeLabel(atom.label);
delete atom.label;
}
}''')
disp.zoomTo()
disp.show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
DONE¶
We encourage you to play around with this tutorial and substitute your own PDB file