Welcome to the EPISOL Colab Playground!

The 3D reference interaction site model (3DRISM) provides an efficient grid-based solvation model to compute the structural and thermodynamic properties of solutes emersed in solvent. However, this solvent need not be aqueous, and can hypothetically be any molecule. In this notebook we will walk through a high-throughput calculation on the solvation free energy of a test set of small molecules, emersed in a common electrolyte solvent.
goals:
utilize an organic solvent
Generate the coordinate and topology files for our test set using RDKit and openFF
Perform high-throughput 3DRISM calculations to determine solvation free energy of 100 electrolyte molecules in the test set
Be able to perform similar calculations on your own molecules
use the colab-notebook to download and run calculations on your own molecules
[ ]:
#@title ##Download and Install Episol
#@markdown ($\approx 2$min) Stable as of 07/01/25 eprism v1.2.6
%%capture
import subprocess
import pandas as pd
import matplotlib.pyplot as plt
#%cd ../home/
%cd $HOME/
%mkdir episol
%cd episol
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/fftw/fftw-3.3.8.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded fftw files"
!echo "+++++++++++++++++++"
!tar -xzf fftw-3.3.8.tar.gz
%cd fftw-3.3.8/
#!./configure --prefix=/home/fftw-3.3.8
!./configure --prefix=$HOME/episol/fftw-3.3.8
!make
!make install
%cd ../
!wget https://github.com/EPISOLrelease/EPISOL/raw/refs/heads/main/src/kernel/release.tar.gz
!echo "+++++++++++++++++++"
!echo "downloaded Episol files"
!echo "+++++++++++++++++++"
!tar -xzf release.tar.gz
%cd release/
#!./configure --with-fftw=/home/fftw-3.3.8
!./configure --with-fftw=$HOME/episol/fftw-3.3.8
!make
!make install
#%cd /content
########################### WRAPEPR
import subprocess
import os
import threading
import pandas as pd
import matplotlib.pyplot as plt
!pip install episol
[ ]:
#@title Install some python packages for topology generation
#@markdown ($\approx$4min)
#@markdown This will prompt a restart in our colab session, this is necessary, just keep moving
#@markdown (if you are using the notebook offline this wont be necessary, as presumably you'll have your own forcefield to generate topologies)
########################################
# FOR COLAB USERS ONLY #
#---------------------------------------#
# if you are running locally you dont need
# condacolab. Just use your local conda dist
########################################
!pip install -q condacolab
import condacolab
condacolab.install()
########################################
#!conda update conda
#!conda install --yes -c conda-forge python=3.11 numpy=1.26.4 openmm pdbfixer parmed mdanalysis py3dmol rdkit openff-toolkit
#!conda install -y -c conda-forge numpy=1.26.4 openmm=8.3.1 python={PYTHON_VERSION} pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 libgcc
#!conda install -y -c conda-forge python=3.12 numpy=1.26.4 openmm=8.3.1 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 torchvision
#openmm pdbfixer parmed mdanalysis py3dmol rdkitconda install libgcc
⏬ Downloading https://github.com/jaimergp/miniforge/releases/download/24.11.2-1_colab/Miniforge3-colab-24.11.2-1_colab-Linux-x86_64.sh...
📦 Installing...
📌 Adjusting configuration...
🩹 Patching environment...
⏲ Done in 0:00:11
🔁 Restarting kernel...
[ ]:
%%capture
!python -m ensurepip --upgrade
!python3.12 -m pip install --upgrade setuptools
#!pip3 install scikit-learn
!conda install -y -c conda-forge python=3.12 numpy=1.26.4 openmm=8.3.1 pdbfixer=1.11 parmed=4.3.0 mdanalysis=2.9.0 py3dmol=2.5.2 rdkit=2025.03.5 openff-toolkit=0.17.0 torchvision pymol-open-source ambertools
[ ]:
#@title import our download packages
%%capture
ias!!ddmport py3Dmol
def MolTo3DView(mol, size=(300, 300), style="stick", surface=False, opacity=0.5):
"""
https://birdlet.github.io/2019/10/02/py3dmol_example/
Draw molecule in 3D
Args:
----
mol: rdMol, molecule to show
size: tuple(int, int), canvas size
style: str, type of drawing molecule
style can be 'line', 'stick', 'sphere', 'carton'
surface, bool, display SAS
opacity, float, opacity of surface, range 0.0-1.0
Return:
----
viewer: py3Dmol.view, a class for constructing embedded 3Dmol.js views in ipython notebooks.
"""
assert style in ('line', 'stick', 'sphere', 'carton')
mblock = Chem.MolToMolBlock(mol)
viewer = py3Dmol.view(width=size[0], height=size[1])
viewer.addModel(mblock, 'mol')
viewer.setStyle({style:{}})
if surface:
viewer.addSurface(py3Dmol.SAS, {'opacity': opacity})
viewer.zoomTo()
return viewer
def smi2conf(smiles):
'''Convert SMILES to rdkit.Mol with 3D coordinates'''
mol = Chem.MolFromSmiles(smiles)
if mol is not None:
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.MMFFOptimizeMolecule(mol, maxIters=200)
return mol
else:
return None
#free_energy("EXP")
!python -m ensurepip --upgrade # since we are using python 3.12 some pkg utils are now obsolete
# after conda-initiate restart colab resets pip
import matplotlib.pyplot as plt
import openmm as mm
from openmm import app
from openmm.unit import *
import py3Dmol as pymol
import MDAnalysis as md
import parmed as chem
from openff.toolkit.topology import Molecule, Topology
import numpy as np
from MDAnalysis.transformations import center_in_box
# from episol import epipy
from rdkit import Chem
from rdkit.Chem import AllChem
from openff.toolkit.topology import Molecule
from openff.toolkit.utils import get_data_file_path
from openff.toolkit.typing.engines.smirnoff import ForceField
from openff.interchange import Interchange
# FIX IMPORTER ERROR
!python -m ensurepip --upgrade
!pip3 install episol
!python3.12 -m pip install --upgrade setuptools
%cd /content/
Procedure¶
To use a solvent besides water, we need bulk solvent-solvent correlations
these can be acquired with MD or with 1DRISM
here we will use a mixture of water and cyanide
[125]:
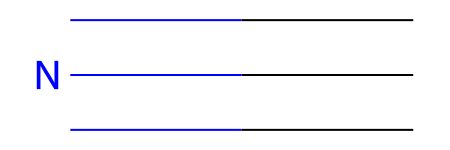
smi = "N#C"
mol = Chem.MolFromSmiles(smi)
mol
[125]:
[126]:
mol = Chem.rdmolops.AddHs(mol,addCoords=True)
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
MolTo3DView(mol).show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
$
h(r)_i^{\rm sol.-solv.} = \rho\sum_j:nbsphinx-math:int `c(r)_j^{:nbsphinx-math:rm sol.-solv.`}:nbsphinx-math:left[omega(r)_j^{rm solv.-solv.} + h(r)_j^{rm solv.-solv.}right]`d:nbsphinx-math:mathbf{r}`$$
$
h(r)_i^{\rm sol.-solv.} \equiv `:nbsphinx-math:exp{left( - U(r)_i^{rm sol.-solv.} + h(r)_i^{rm sol.-solv.}+ c(r)_i^{rm sol.-solv.} + mathfrak{B}right)}` $$
we will download our correlations here
[48]:
%%capture
!wget https://raw.githubusercontent.com/p-swanson/solvents/refs/heads/main/organic/cyanide.gvv
!wget https://raw.githubusercontent.com/p-swanson/solvents/refs/heads/main/organic/cyanide.sol
we can see that we have a mixture of cyanide and water.
this correspondings to 5 sites in total
here we will test our system with the classic methane example
[ ]:
!wget https://raw.githubusercontent.com/p-swanson/solvents/refs/heads/main/tests/methane.gro
!wget https://raw.githubusercontent.com/p-swanson/solvents/refs/heads/main/tests/methane.top
[123]:
test = int()
[124]:
mem = epipy("methane.gro","methane.top",gen_idc=True)
mem.closure = "KH"
mem.err_tol = 1e-04
mem.ndiis = 15
mem.solvent("cyanide.sol")
mem.solvent_path = "./"
mem.report(f"methane_cyanide_{mem.closure}")
mem.rism(step=2000,resolution=0.5)
mem.kernel(nt=2)
test +=1
converted methane.top to methane.solute
generated idc-enabled solute file to: idc_methane.solute
Calculation finished in 42 steps
err_tol: 0.0001 actual: 9.96146e-05
[53]:
guv = mem.select_grid()
[60]:
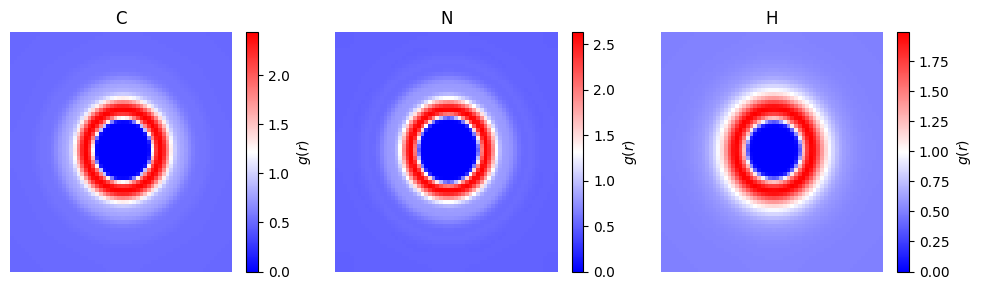
SLICE = 30
names = ["C","N","H"]
fig,axs = plt.subplots(1,3,figsize=(10,3))
for ind,(ax,name) in enumerate(zip(axs.flatten(),names)):
# select values
t = mem.reader(f"methane_cyanide_{mem.closure}.txt",atom_to_select=ind+3)
p = ax.pcolormesh(t[SLICE],cmap='bwr')
ax.set_title(name)
fig.colorbar(p,ax=ax,label="$g(r)$")
ax.set_axis_off()
fig.tight_layout()